
Fabry Disease:
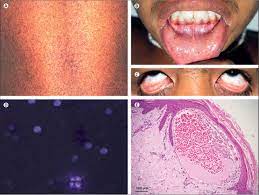
The Lysosomal Storage Disorders Support Society sought the Union Ministry of Health and Family Welfare’s immediate intervention for the treatment of Fabry Disease patients.
- Fabry Disease is a rare inherited disorder of glycosphingolipid (fat) metabolism resulting from the absent or markedly deficient activity of the lysosomal enzyme, alpha-galactosidase A (α-Gal A).
- It belongs to a group of diseases known as lysosomal storage disorders.
- This enzymatic deficiency is caused by alterations (mutations) in the α-galactosidase A (GLA) gene that instructs cells to make the α-galactosidase A (α-Gal A) enzyme.
- Lysosomes function as the primary digestive tract of cells.
- Symptoms: Numbness, tingling, burning or pain in the hands or feet, extreme pain during physical activity and heat or cold intolerance etc.
- The first indication of a problem may be kidney failure or heart disease.
- The patients are treated by intravenously administered enzyme replacement therapy (ERT) or Oral Chaperone Therapy


